
甘肃省药品监督管理局关于岷归道地药材质量标准的公告
甘肃省药品监督管理局关于岷归道地药材质量标准的公告

参照《甘肃省中藏药材地方标准审定发布工作规范(试行)》,岷归道地药材标准经省药监局组织专家技术审评通过,符合甘肃省中药材标准要求,现予以公告。
附件:岷归药材质量标准。
前 言
本标准在遵从《中华人民共和国药典》的基础上,提出“岷归”道地药材的质量标准。
实验样品采自甘肃省各地的当归产区,同时采集了省外有关产区的当归样品,研究制定了“岷归”
道地药材的质量标准。
本标准由甘肃省药品监督管理局提出并归口。
本标准负责起草单位:甘肃省药品检验研究院、陇西保和堂药业有限责任公司、岷县当归研究院。
本标准参加起草单位:中国食品药品检定研究院岷县中药材生产技术指导站、甘肃省道地药材研
究所、甘肃省中藏药检验检测技术工程研究中心、甘肃省中藏药检验检测技术工程实验室、甘肃省药学会。
本标准主要起草人:杨平荣、宋平顺、倪琳、魏锋、马双成、马中森、李开银、贺军权、马潇。
本标准参加起草人:吴仲涛、石鹏刚、郭增祥、季贵文、郎建军、刘富强、刘志浩、杨耘、卢雪蕊、蔺瑞丽、王亚飞、刘昭伟、唐想芳、李文义。
本标准为首次发布,为推荐性标准。
岷归道地药材质量标准
1 范围
本标准规定了岷归道地药材的术语、定义、来源、道地产区、质量特征
本标准适用于岷归道地药材的生产、销售和使用的质量检验。
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅所注日期的版本适用于本文件。凡是不注日期的引用文件,其最新版本(包括所有的修改单项)适用于本文件。
中华人民共和国药典(2015 年版一部、四部)
3 术语和定义
下列术语和定义适用于本标准。
3.1当归
本品为伞形科植物当归Angelica sinensis(Oliv)Diels的干燥根。
主产于甘肃,四川、云南、青海亦产。为甘肃的道地药材。
3.2 岷当归 Min danggui
产自甘肃省岷县及周边地区的当归道地药材,简称岷归。
3.3岷当归道地产区
岷当归道地产区,位于甘肃南部的岷山山脉东支南北两面山麓地区和陇中黄土高原与青藏高原的交汇过渡带,包括甘肃的岷县、宕昌、临潭、卓尼和漳县等,海拔2400~2800m的高寒二阴地区。
4.技术要求
4.1感官要求
应符合表1的规定。

注:柴性大、干枯无油或断面呈绿褐色者不可供药用。
4.2显微鉴别要求
应符合表2的规定。
表2 显微鉴别要求

4.3理化指标
应符合表3的规定。


4.4重金属及有害元素
表3 重金属及有害元素

4.5 二氧化硫残留量
表5 二氧化硫残留量

5检验方法
5.1感官要求
按《中国药典》2015年版一部当归项下规定的【性状】方法检验。
5.2显微鉴别要求
按《中华国药典》2015年版一部当归项下规定的【鉴别】(1)方法检验。
6.附注
当归药材中的农药残留量,照农药残留量测定法(中国药典2015年版四部通则2341)测定。
附录A
特征图谱鉴别
1 范围
本标准是采用高效液相色谱仪进行当归药材和饮片中掺假独活、掺假欧当归分析的一般规定。
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅所注日期的本适用于本文件。凡是不注日期的引用文件,其最新版本(包括所有的修改单项)适用于本文件。
中华人民共和国药典(2015 年版一部、四部)
国家药品标准技术规范(2013年第4版)
3.术语和定义
3.1特征图谱
是指中药经过适当的处理后,采用一定的分析方法得到的能够标识其中重要特征信息的图谱。
3.2 掺假
在正品药材或饮片中非法掺入外观性状相似的非同种类物质的行为。
3.2.1 掺假独活
在当归饮片中非法掺入独活饮片(重齿毛当归Angelica pubescens Maxim.f.biserrata Shan et Yuan的干燥根的切制品)。
3.2.2掺假欧当归
在当归饮片中非法掺入欧当归饮片(欧当归Levisticum officinale Koch.的干燥根的切制品)。
4.试验方法
照高效液相色谱法(中国药典2015年版通则0512)测定。
4.1色谱条件
以十八烷基硅烷键合硅胶为填充剂;以乙腈流动相A,1.0%乙酸酸溶液为流动相B,按表进行梯度洗脱。柱温为室温。检测波长为280nm。理论板数按阿魏酸峰计算应不低于5000。 4.2参照物溶液的制备 取阿魏酸对照品适量,精密称定,置棕色量瓶中,加70%甲醇制成每1ml含阿魏酸12µg的溶液,摇匀,即得。
4.2参照物溶液的制备 取阿魏酸对照品适量,精密称定,置棕色量瓶中,加70%甲醇制成每1ml含阿魏酸12µg的溶液,摇匀,即得。
4.3供试品溶液的制备 取样品粉末(过三号筛)约0.2g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理40分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,静置,取上清液滤过,取续滤液,即得。
4.4测定法 分别精密吸取参照物溶液与供试品溶液各20µl,注入液相色谱仪,测定,记录100分钟的色谱图,即得。
4.5特征图谱 供试品特征图谱中应呈现5个特征峰,与参照物峰相对应的峰为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间应在规定值的±5%之内,规定值为:0.60(峰1)、0.65(峰2)、 1.0(峰S)、 2.26(峰3) 和2.35(峰4)。
4.5.1特征图谱鉴别(1)(特异色谱峰检查供试品中掺假独活)
在供试品特征图谱中,不应出现2个特异色谱峰,与阿魏酸峰(S峰)的相对保留时间分别为1.57±5%和2.41±10%。
4.5.2特征图谱鉴别(2) (比值法检查供试品中掺假欧当归)
在供试品特征图谱中,在与阿魏酸峰(S峰)的相对保留时间分别为0.66±5%(峰1)和1.19±5%(峰2)出现2个色谱峰,其峰1和峰2峰面积之和与样品中阿魏酸峰面积的比值应不得过1.0。

图 1 岷当归药材特征图谱(S:阿魏酸)
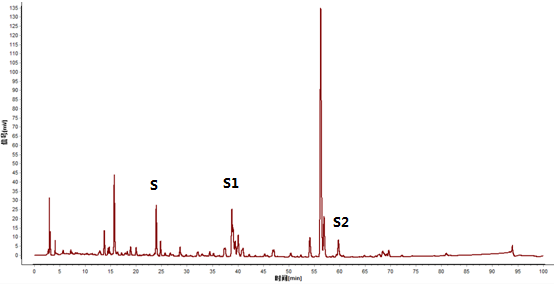
图 2 独活药材特征图谱(S:阿魏酸)
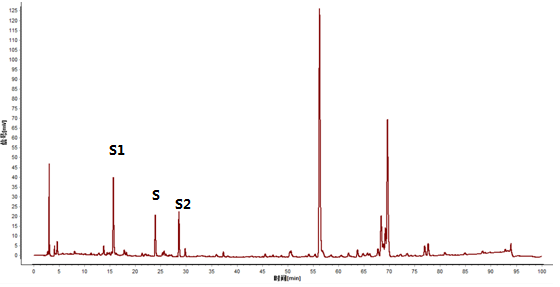
图 3 欧当归指纹图谱(S:阿魏酸)
附录B
红外光谱鉴别
1 范围
本标准是采用傅里叶变换红外光谱仪进行当归药材的定性分析及相关性分析。
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅所注日期的本适用于本文件。凡是不注日期的引用文件,其最新版本(包括所有的修改单项)适用于本文件。
中华人民共和国药典(2015年版四部)
ATC 009 红外光谱分析技术
GB/T 14666-1993 分析化学
GB/T 7764-2001 橡胶鉴定 红外光谱法
GB/T 6040-2002 红外光谱分析方法通则
3 方法依据
中药样品含有非常复杂的化学组分,所含基团及其数量各不相同,分子结构各不相同,当红外辐射通过液体或固体中药样品时,将在不同波长处出现它们各自特有的吸收峰(带)强度的叠加,以波长或波数为横坐标,吸光度或透过率为纵坐标绘图,得到样品的特征吸收曲线为红外吸收光谱。以红外光谱的宏观形貌以及吸收峰的峰位置、峰强度和峰形状整体指纹特征来定性判断或鉴别样品之用。以某些特征谱带或特征峰的相对强度来确定样品中某组分的定性分析之用。
4 仪器和设备
傅里叶变换红外光谱仪: 仪器分辨率优于4cm-1,基线噪声6000:1~2000:1,波数范围4000~400cm-1,基线倾斜0.1%T~0.5%T,透过率准确度0.1%T,透过率重复性0.1%T~0.5%T,吸光度重复性<0.005。
5 安装环境条件
5.1仪器应安装在清洁无尘、无震动、无电磁干扰、无腐蚀性气体的实验室内。
5.2应尽量避免日光的直接照射,室内温度应控制在20~25oC范围内,湿度<60%。
5.3供给电源要有稳压设备,使电压稳定在220±5V,50Hz。
6.试样和试剂
6.1 试样
为产于全国各地的当归药材样品。
6.2 试剂
优级纯或光谱纯的溴化钾(KBr)。
6.3标准物
对照药材由国务院药品监督管理部门指定的单位制备、标定和供应。
7.分析步骤
7.1 样品制备
7.1.1对照中药材试样
按照中国药典取样方法取样,粉碎后过200目(74μm)筛的粉末直接用KBr压片法测定。
7.1.2中药材试样
按照中国药典取样方法取样,粉碎后过200目(74μm)筛的粉末直接用KBr压片法测定。
7.1.3 KBr压片法(红外烤灯下操作)
取本品粉末(过 200 目)约 3mg, KBr 碎晶 150mg,置于玛瑙研钵中,磨细,并使其混合均匀。将研细混匀的粉末倾入 13mm 压片模具中,使其铺布均匀,加压至 0.8GPa~1GPa,保持 1min,使用压片机将其压成圆形薄片,另取约 3mg 的当归对照药材粉末(过 200 目),同法操作,压成圆形薄片,然后将供试品、对照药材薄片放入红外光谱仪进行测试,记录红外光谱图。在扫描待测样品前,先用纯KBr按前述方法压制成透明薄片,按照与扫描待测样品相同的参数设置方式进行测试,得到光谱背景。接下来扫描待测样品的光谱时,仪器会自动进行背景扣除,得到待测样品的红外光谱。
7.2测定条件
7.2.1 仪器的参数
扫描范围:4000~400cm-1(波长2.5μm~25μm)
分辨率:4cm-1
扫描次数:16~32次,当噪音大时可适当增加次数。
7.2.2 制样时试样和KBr的用量
试样对KBr的比例,正常情况下一般试样占1~2%(质量百分数),以所得红外谱图中基线与最强吸收峰的透过率差值(Y基线-Y最强峰)≥70%为宜。高含油酯、蛋白类等特殊试样其透过率差值会适当减小。
7.2.3 试样的重复测定次数
中药材: 重复3次 相关系数在≥0.98为宜。
8.定性分析
取3mg的样品与150mg的KBr碎晶置于玛瑙研钵中,磨细,并使其混合均匀。将研细混匀的粉末倾入13mm压片模具中, 使其铺布均匀,加压至0.8~1 GPa,保持1min,使用压片机将其压成圆形薄片,另取对照药材粉末3mg, 同法操作,压成圆形薄片,然后分别将样品和对照药材压片放入红外光谱仪进行测试,记录光谱图。通过供试品红外光谱与当归红外光谱标准普库相似度匹配,进行定性分析及相关性分析。

当归对照药材红外光谱图








 甘公网安备 62112602000025号
甘公网安备 62112602000025号